Quickstart - calling methylation with nanopolish¶
Oxford Nanopore sequencers are sensitive to base modifications. Here we provide a step-by-step tutorial to help you get started with detecting base modifications using nanopolish.
For more information about our approach:
- Simpson, Jared T., et al. “Detecting DNA cytosine methylation using nanopore sequencing.” Nature Methods (2017).
Requirements:
Download example dataset¶
In this tutorial we will use a subset of the NA12878 WGS Consortium data. You can download the example dataset we will use here (warning: the file is about 2GB):
wget http://s3.climb.ac.uk/nanopolish_tutorial/methylation_example.tar.gz
tar -xvf methylation_example.tar.gz
cd methylation_example
Details:
- Sample : Human cell line (NA12878)
- Basecaller : Guppy v2.3.5
- Region: chr20:5,000,000-10,000,000
In the extracted example data you should find the following files:
albacore_output.fastq: the subset of the basecalled readsreference.fasta: the chromsome 20 reference sequencefast5_files/: a directory containing signal-level FAST5 files
The reads were basecalled using this albacore command:
guppy_basecaller -c dna_r9.4.1_450bps_flipflop.cfg -i fast5_files/ -s basecalled/
After the basecaller finished, we merged all of the fastq files together into a single file:
cat basecalled/*.fastq > output.fastq
Data preprocessing¶
nanopolish needs access to the signal-level data measured by the nanopore sequencer. To begin, we need to create an index file that links read ids with their signal-level data in the FAST5 files:
nanopolish index -d fast5_files/ output.fastq
We get the following files: output.fastq.index, output.fastq.index.fai, output.fastq.fastq.index.gzi, and output.fastq.index.readdb.
Aligning reads to the reference genome¶
Next, we need to align the basecalled reads to the reference genome. We use minimap2 as it is fast enough to map reads to the human genome. In this example we’ll pipe the output directly into samtools sort to get a sorted bam file:
minimap2 -a -x map-ont reference.fasta output.fastq | samtools sort -T tmp -o output.sorted.bam
samtools index output.sorted.bam
Calling methylation¶
Now we’re ready to use nanopolish to detect methylated bases (in this case 5-methylcytosine in a CpG context). The command is fairly straightforward - we have to tell it what reads to use (output.fastq), where the alignments are (output.sorted.bam), the reference genome (reference.fasta) and what region of the genome we’re interested in (chr20:5,000,000-10,000,000):
nanopolish call-methylation -t 8 -r output.fastq -b output.sorted.bam -g reference.fasta -w "chr20:5,000,000-10,000,000" > methylation_calls.tsv
The output file contains a lot of information including the position of the CG dinucleotide on the reference genome, the ID of the read that was used to make the call, and the log-likelihood ratio calculated by our model:
chromosome start end read_name log_lik_ratio log_lik_methylated log_lik_unmethylated num_calling_strands num_cpgs sequence
chr20 4980553 4980553 c1e202f4-e8f9-4eb8-b9a6-d79e6fab1e9a 3.70 -167.47 -171.17 1 1 TGAGACGGGGT
chr20 4980599 4980599 c1e202f4-e8f9-4eb8-b9a6-d79e6fab1e9a 2.64 -98.87 -101.51 1 1 AATCTCGGCTC
chr20 4980616 4980616 c1e202f4-e8f9-4eb8-b9a6-d79e6fab1e9a -0.61 -95.35 -94.75 1 1 ACCTCCGCCTC
chr20 4980690 4980690 c1e202f4-e8f9-4eb8-b9a6-d79e6fab1e9a -2.99 -99.58 -96.59 1 1 ACACCCGGCTA
chr20 4980780 4980780 c1e202f4-e8f9-4eb8-b9a6-d79e6fab1e9a 5.27 -135.45 -140.72 1 1 CACCTCGGCCT
chr20 4980807 4980807 c1e202f4-e8f9-4eb8-b9a6-d79e6fab1e9a -2.95 -89.20 -86.26 1 1 ATTACCGGTGT
chr20 4980820 4980822 c1e202f4-e8f9-4eb8-b9a6-d79e6fab1e9a 7.47 -90.63 -98.10 1 2 GCCACCGCGCCCA
chr20 4980899 4980901 c1e202f4-e8f9-4eb8-b9a6-d79e6fab1e9a 3.17 -96.40 -99.57 1 2 GTATACGCGTTCC
chr20 4980955 4980955 c1e202f4-e8f9-4eb8-b9a6-d79e6fab1e9a 0.33 -92.14 -92.47 1 1 AGTCCCGATAT
A positive value in the log_lik_ratio column indicates support for methylation. We have provided a helper script that can be used to calculate how often each reference position was methylated:
scripts/calculate_methylation_frequency.py methylation_calls.tsv > methylation_frequency.tsv
The output is another tab-separated file, this time summarized by genomic position:
chromosome start end num_cpgs_in_group called_sites called_sites_methylated methylated_frequency group_sequence
chr20 5036763 5036763 1 21 20 0.952 split-group
chr20 5036770 5036770 1 21 20 0.952 split-group
chr20 5036780 5036780 1 21 20 0.952 split-group
chr20 5037173 5037173 1 13 5 0.385 AAGGACGTTAT
In the example data set we have also included bisulfite data from ENCODE for the same region of chromosome 20. We can use the included compare_methylation.py helper script to do a quick comparison between the nanopolish methylation output and bisulfite:
python3 compare_methylation.py bisulfite.ENCFF835NTC.example.tsv methylation_frequency.tsv > bisulfite_vs_nanopolish.tsv
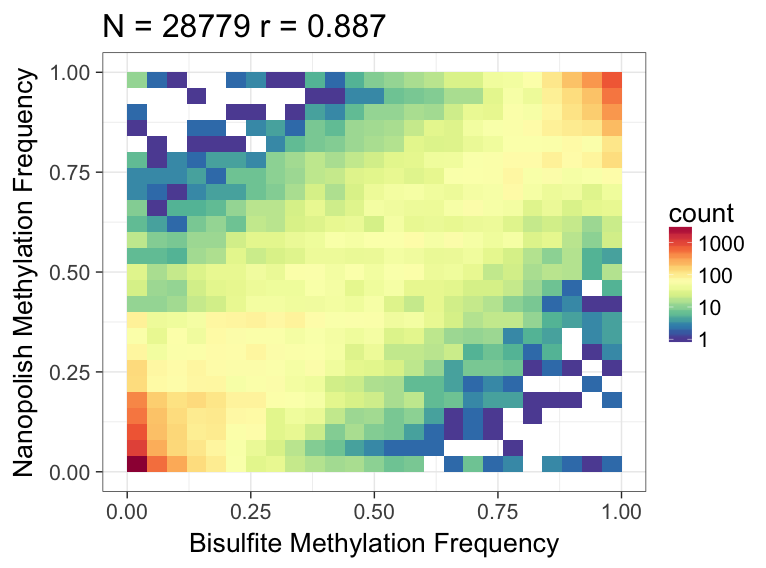
We can use R to visualize the results - we observe good correlation between the nanopolish methylation calls and bisulfite:
library(ggplot2)
library(RColorBrewer)
data <- read.table("bisulfite_vs_nanopolish.tsv", header=T)
# Set color palette for 2D heatmap
rf <- colorRampPalette(rev(brewer.pal(11,'Spectral')))
r <- rf(32)
c <- cor(data$frequency_1, data$frequency_2)
title <- sprintf("N = %d r = %.3f", nrow(data), c)
ggplot(data, aes(frequency_1, frequency_2)) +
geom_bin2d(bins=25) + scale_fill_gradientn(colors=r, trans="log10") +
xlab("Bisulfite Methylation Frequency") +
ylab("Nanopolish Methylation Frequency") +
theme_bw(base_size=20) +
ggtitle(title)
Here’s what the output should look like: